Introduction
Over the past decade, significant advances have improved our understanding of the genetic and molecular mechanisms that lead to cancer. And yet, a recent review of the oncology drugs approved by the U.S. Food and Drug Administration (FDA) on the basis of improvement in tumor response rates showed only 10 percent of these therapies demonstrate an overall survival benefit.1
With an etiology that involves an array of genetic interactions and dysfunction across multiple systems, cancer is one of the most scientifically complex and dynamic diseases. This complexity makes the design of oncology clinical trials, especially early-stage studies, challenging.
With the emergence of precision medicine, we are seeing a shift in how early-phase oncology trials are conducted, including a growing number of Phase 1 trials reporting preliminary tumor response rates. This shift is due in part to an increase in adaptive trial designs that seek to limit the number of patients exposed to ineffective doses or treatments while accelerating the timeline to the detection of efficacy signals.
In this white paper, we address clinical trials in precision medicine and explore the expanding role of adaptive trial designs in Phase 1 and Phase 2 oncology studies.

About precision medicine
The definition of precision medicine varies from source to source. This paper adopts the definition that precision medicine is an approach to medicinal practice that tailors treatment decisions to a patient’s individual characteristics, which may be genetic, anatomical, physiological, environmental, or lifestyle-related. The overarching goal of precision medicine is to account for the systemic variability of individuals within a population when selecting appropriate therapies.
To that end, clinical trials investigating targeted therapies often involve the evaluation of key biomarker or biological analytes that inform the therapy’s effectiveness. In fact, the FDA, the European Medicines Agency (EMA), and other regulatory authorities may require the development of a companion diagnostic for drug approval.
As sponsors seek to develop targeted therapeutics, it is important to consider how study design may impact the efficiency and flexibility of their trials.
Current state of precision medicine in oncology
When thinking about biomarkers or particular characteristics of a patient population, it is important for sponsors to consider where they are in the drug development life cycle. Limiting the eligible study population based on specific biomarkers could negatively impact the enrollment rate for a trial. However, the use of biomarkers can also help sponsors select a population that is more likely to have non-confounded safety readouts while also improving the potential to see an efficacy signal where one is expected based on preclinical research.
Generally, the objectives of Phase 1 studies are dose selection, safety assessment, and development of an initial safety profile. Phase 2 studies are typically designed to give an initial readout of an efficacy signal. However, there are special considerations in oncology trials where Phase 1 studies may be driven by the mechanism of action and include patients with different tumor types.
In addition to dose selection and safety assessment, a goal of these Phase 1 studies is to identify the indication(s) to be pursued in Phase 2 studies. In each of these early-phase studies, biomarkers can be used to enrich the study population by informing the selection of patients who are most likely to tolerate and to respond to treatment, thereby limiting the number of patients who receive ineffective therapy or doses and minimizing the likelihood of early-stage product failure due to inappropriate patient selection.
It is important for sponsors to keep in mind that biomarkers are not limited to tumor mutations or genomic variations and can also include biological variables such as the Eastern Cooperative Oncology Group (ECOG) Performance Status, which measures functional status. As an example, higher ECOG scores as eligibility criteria for short Phase 1 studies may be useful to augment enrollment, whereas lower ECOG scores will help select those patients who are more likely to tolerate extended treatment periods in longer Phase 2 studies.

Rationale for adaptive trial designs
Fixed trial designs follow three key steps – design, conduct, and analysis – and require waiting until the end of the study to look at the data and draw any conclusions. Adaptive design trials offer additional flexibility by allowing the review of data at prespecified times during study conduct and the ability to make predefined adaptations to study design elements based on observations that are accruing within the trial.
The key is to ensure that all planned adaptations are defined a priori during trial design and before the trial begins. This avoids the perception of bias where adaptations to the trial are decided upon only after observation of ongoing trial data. Adaptive designs do not necessarily need to be complicated. A standard 3+3 dose-escalation design with a review of dosing in between cohorts is an adaptive design trial in which a committee is charged with deciding whether to dose-escalate, stop dosing, stop the trial, or even go to an intermediate dose based on data observed thus far. The inclusion of an interim analysis can also be considered an adaptive design.
In precision medicine oncology trials, design adaptations are often based on predictive or prognostic genetic biomarkers:
- Predictive biomarkers, which predict the likelihood of response to a specific therapy, are used for allocating individual patients to suitable treatment arms. Examples of predictive biomarkers include K-ras mutation status in colorectal cancer and epidermal growth factor receptor mutation status in early-stage lung cancer.
- Prognostic biomarkers, which predict the most likely prognosis of an individual patient, are useful for providing valid comparisons between treatments. Examples of prognostic biomarkers include tumor mutation burden, tumor size, presence of metastasis, and number of positive lymph nodes.
Adaptive design trials offer many advantages over conventional fixed-sample design trials. As long as the a priori assumptions and design are accepted by regulatory authorities, an adaptive design approach minimizes the need for amendments or new protocols, which can be costly and require retraining of the sites. For adaptive design trials, regulatory guidance stipulates specification of pre-planned changes and the intended analysis in the study protocol to maintain study integrity and validity. Pre-planned changes may include:2
- Refinements in sample size
- Dynamic adjustment of dose schedules, including dropping or adding treatments or doses
- Changes in treatment-arm allocations
- Narrowing the population to those patients most likely to benefit from the treatment
- Early stopping decisions
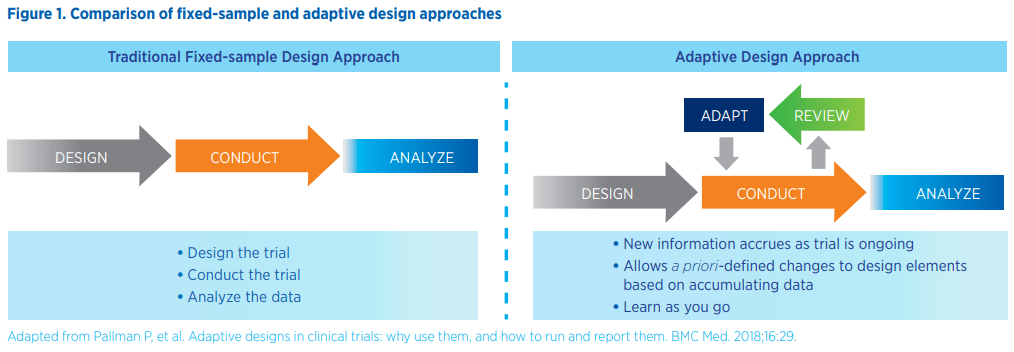
Adaptive design approaches also enable seamless transition between study phases by eliminating the requirement to close down a trial after performing a final analysis and before opening a new trial protocol. These trials may be more efficient since sites contributing to the first phase will remain open, eliminating the necessity for study start-up activities at these sites and potentially requiring fewer participants. Such trials may also give patients the opportunity to continue therapy in the same trial until progression rather than re-enroll in a rollover study or obtain the drug through expanded access programs. Importantly, the flexibility inherent in adaptive design trials may reduce the number of patients exposed to ineffective treatments or doses and the time needed to make informed decisions about treatment safety and efficacy, especially in the early stages of clinical development. For example, an accelerated 3+3 design or a continuous reassessment model for dose escalation may enable quicker identification of the maximum tolerated dose (MTD) or minimal effective dose than a traditional 3+3 design. At the same time, adaptive designs allow for an early efficacy readout based on a priori criteria established for multiple planned interim looks.
Early-phase oncology studies that include patients with a common mutation but different solid tumors, known as basket trials, are a commonly used adaptive design that allows sponsors to allocate cohorts of patients to specific histologies that are most promising and to discontinue cohorts not showing efficacy, thereby limiting risk to the overall treatment development plan. Finally, given the trend toward combination therapies in precision medicine oncology trials, adaptive designs may allow for dose finding and dose expansion with add-on drugs, all within the same trial design construct.
Design considerations for precision medicine oncology trials
As sponsors prepare to design an early-stage oncology study, they will need to:
- Define the trial objective, whether it is safety, pharmacology activity, preliminary tumor response signal, feasibility assessment, or a combination of these
- Define the study population and subgroups with special consideration of how to handle patients who progress after initial treatment, especially if using overall survival as an endpoint
- Define the methodology used to assess response, given that biomarkers may be more sensitive than tumor measurements in gauging response, particularly for targeted therapies and immunotherapies
Operational considerations with adaptive trials
The successful execution of adaptive trial designs requires a cross-functional effort and careful consideration of critical operational elements. The first among these elements is choosing whether to include a control group and whether or not to blind the study. Selection of an appropriate control group facilitates the interpretation of safety data and assessment of early activity or efficacy. Where possible, using a standard-of-care control is preferred to a placebo control, especially in disease areas like oncology, where it may be unethical to assign patients to placebo alone. Also, sponsors should consider using historical controls when concurrent controls are not feasible.
The second operational element to consider is dose and regimen. When implementing adaptive designs in Phase 1 studies, sponsors will need to decide what the starting dose should be, how many doses to include, and what the dose-escalation strategy will be. This is important because, in some model-based designs such as the modified total probability interval model, efficiency increases as the number of doses increases.
The third operational element sponsors will need to think through is the treatment plan itself, including cohort size and cohort management. Cohort size is particularly important in dose escalation studies for limiting the number of patients exposed to ineffective doses and in dose expansion to enable a reasonable estimation of efficacy.
Common adaptive designs for precision medicine trials in oncology
Adaptive design approaches that can be used to optimize early- stage oncology trials in precision medicine include:
- Seamless Phase 1/2 adaptive designs, where a Phase 1 study targeted at finding the MTD transitions into a Phase 2 expansion study aimed at determining an efficacy signal
- Biomarker enrichment designs, which use biomarkers to enrich the study population with patients who are more likely to respond to treatment
- Biomarker stratified designs, in which biomarkers are measured on all patients and used as stratification variables
- Umbrella designs, which evaluate multiple targeted therapies for a single disease that is stratified into subgroups by molecular alterations
- Basket designs, in which a targeted therapy is evaluated on multiple diseases that have common underlying molecular alterations
As can be seen above, adaptive design studies may include multiple cohorts and multiple tumor types. It is also important to keep in mind that numerous adaptation methods may be used in a single trial and may facilitate a more rapid, seamless transition between study phases.3
Case study: seamless Phase 1/2 adaptive design
In this case study, a new PD1 inhibitor, “XYZ,” is being investigated as both a monotherapy and as a combination therapy with standard of care. The study design includes all comers in three cancer populations – non-small-cell lung cancer, prostate cancer, and colorectal cancer. The trial begins with a Phase 1 dose escalation study to determine the MTD. The target dose-limiting toxicity is 30 percent, and four doses will be tested, with subjects receiving three weeks of treatment at each dose. The design employs a Bayesian model-based approach, using the Bayesian Optiman INterval Model (BOIN), which is efficient at identifying the MTD and, subsequently, the recommended Phase 2 dose (RP2D). The trial begins with implementing BOIN in the dose-escalation phase to find the MTD and determine the RP2D on monotherapy.
This RP2D is then investigated further in the Phase 2 dose expansion, using a basket-study design for each of the three cancer populations, aimed at evaluating the drug’s efficacy as a monotherapy for the respective cancer types. Each basket study takes the form of a two-stage adaptive design to detect an efficacy signal. The first stage uses only a small number of patients to evaluate the futility of the monotherapy dose. If the treatment is deemed futile, the trial stops with no further patient exposure to the drug; otherwise, the trial continues to enroll more patients into the second stage, at the end of which evaluation for an efficacy signal is carried out. This two-stage approach limits the number of patients potentially exposed to an ineffective drug.
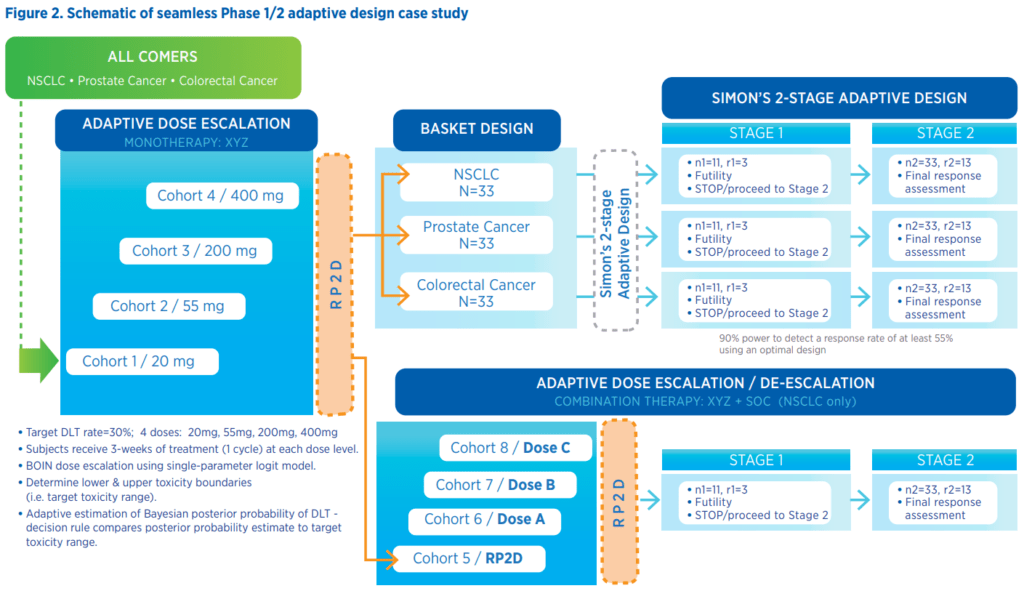
In parallel with the monotherapy expansion study, the RP2D is also used as a starting dose in a second dose-escalation design to determine the MTD under the combination treatment regimen, XYZ combined with standard of care. The resulting new RP2D of the combination regimen is expanded into a corresponding two-stage design to evaluate futility and detect an efficacy signal on the combination treatment regimen, as described earlier.
The benefits of this type of adaptive trial design in the early- phase setting include the ability to stop the study early for futility, which can help limit patient exposure to ineffective treatments. In addition, if an efficacy signal is detected for prostate cancer (as an example) but not the other two disease states, those disease cohorts can be stopped without any need for a protocol amendment.
Challenges with adaptive designs
Despite the inherent benefits of adaptive design in oncology trials, there are still hurdles that may need to be addressed when executing these studies. For example, adaptive trials pose logistical challenges related to implementing the necessary data monitoring and data management processes required by each adaptation. It is also possible that scientific constraints may limit expected efficiency gains from adaptations. In general, when drafting an adaptive design protocol, it is prudent to invest upfront in necessary resources and time for planning the design strategy, and it is particularly important to consult a suitably experienced biostatistician.
Key Takeaway
A recent study carried out by the Biotechnology Innovation Organization found that nearly one-third of drugs entering Phase 2 studies between 2006 and 2015 failed to progress.4 The use of appropriate adaptive design at the early stages of oncology drug development may enable more rapid determinations of futility and more informed predictions of success in later-stage studies, accelerating the overall timeline for the development of novel cancer treatments.
References
[1] Chen EY, Raghunathan V, Prasad V. An overview of cancer drugs approved by the U.S. Food and Drug Administration based on the surrogate end point of response rate. JAMA Intern Med. 2019;179(7):915-921.
[2] Pallman P, et al. Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Med. 2018;16:29.
[3] Sverdlov O, Wong WK. Novel statistical designs for Phase I/II and Phase II clinical trials with dose-finding objectives. Ther Innov Regul Sci. 2014;48:601–612.
[4] Thomas DW, et al. Clinical development success rates 2006-2015. Biotechnology Innovation Organization, Washington D.C. June 2016. Available at: https://www.bio.org/sites/default/ files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20 Biomedtracker,%20Amplion%202016.pdf. Accessed March 25, 2020.