Developing a new biopharmaceutical product is a lengthy, high-stakes journey. It takes, on average, at least 10 years and over $2 billion to successfully bring a new drug to market, and only 10% to 15% of products ultimately receive regulatory approval.[1]
A comprehensive plan and the right regulatory and therapeutic expertise can significantly accelerate the development timeline and increase the likelihood of marketing success, especially for small biotech and specialty pharma companies working with limited time and resources. This post provides an overview of the critical planning activities and milestones in the drug development process, along with important regulatory considerations at each stage.
Stage 1: Planning
Planning is the bedrock of success and involves a lengthy checklist of must-dos. Perfect planning is not critical, but a lack of any planning will certainly jeopardize achieving one’s goals. With a plan in place at the outset, you can make adjustments at each phase of the program as needed.
To optimize a drug development program for a new therapeutic entity, thoroughly think through the following steps:
- Establish program goals
- Define the regulatory strategy
- Clarify critical requirements for approval
- Outline the product development plan at the beginning
The drug development plan should include critical product information, such as:
- Indication and usage statement describing the targeted disease or condition
- Intended usage definition: for treatment, prevention, mitigation, cure, relief, or diagnosis
- Dosage form and route of delivery
- Efficacy expectations measured by defined outcomes or endpoints
- Safety expectations
Also essential in the planning phase is determining the geographic scope of commercialization and the applicable regulatory guidelines. Thoroughly review these commercial considerations early on for strategic market planning:
- Are competitive products already available?
- How is the new drug different from existing treatment options?
- What is the potential market share for the investigative drug?
- Can the product qualify for a special designation by the U.S. Food and Drug Administration or other regulatory body based on its potential to address unmet medical needs or a condition for which treatments either do not exist or are inadequate?
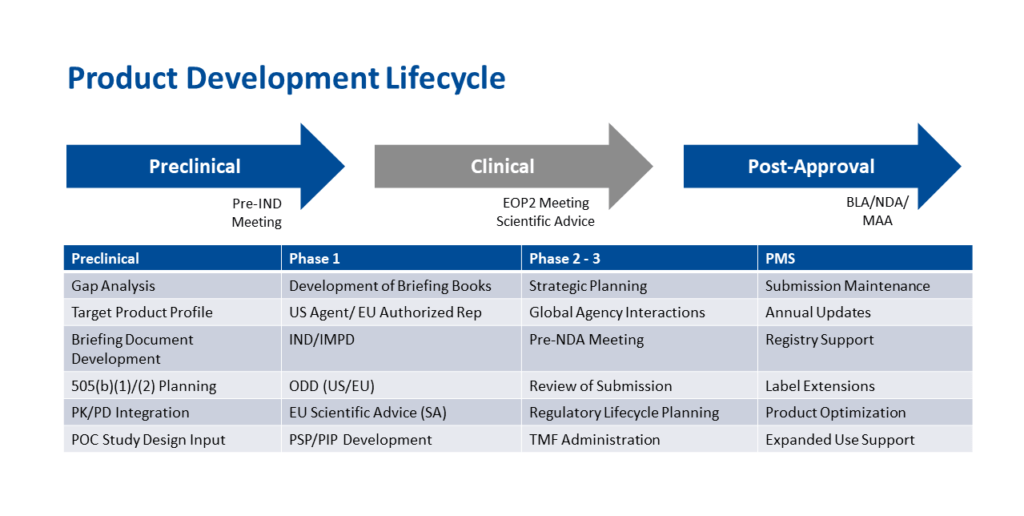
Stage 2: Preclinical Development
The first step in evaluating a new chemical entity or new molecular entity is conducting in vitro and then in vivo preclinical studies that define relevant safety characteristics.
- Safety pharmacology studies assess how the drug affects the respiratory, cardiovascular, and central nervous systems, among others
- Pharmacodynamics studies establish the drug’s mechanism of action and contribute to dose selection for clinical studies
- Toxicology studies define how much of the drug can be safely administered and how frequently, including:
- The maximum tolerated dose
- The no-observed-adverse-effect level, which determines the first-in-human dosage
- Whether repeat doses are toxic, for example with chronic use in treating diabetes or hypercholesterolemia
- Pharmacokinetics assays characterize how the drug is absorbed, distributed, metabolized, and excreted
Further testing for carcinogenicity, immunotoxicity, drug-drug interactions, and other effects continues during clinical development.
Chemistry, manufacturing, and controls planning at the preclinical stage assures that manufacture of the drug is carefully considered at all stages, from development batches through commercial submission. This includes:
- Defining active ingredients and raw materials
- Developing and scaling up the manufacturing process
- Setting in-process controls
- Developing and qualifying analytical methods
- Determining stability requirements
- Establishing guidance on setting and justifying product specifications
Clinical trial design is the final step of the preclinical phase. Scientists determine the appropriate amount of the drug to achieve both safety and efficacy through microdose, single ascending dose, or multiple ascending dose trials. For randomized trials, decisions must be made on the type of blinding. Will the new entity be tested against an approved comparator or placebo? Supplemental safety studies may also be required at one or more phases.
When the trial design has been finalized, an investigational new drug (IND) or analogous clinical trial application must be filed with the FDA, the European Medicines Agency, and/or the regulatory bodies of any other countries where you are planning to conduct the study. This application requests authorization to administer the biopharmaceutical to humans. Requesting a pre-IND meeting with the agency prior to submission may be useful for resolving questions, addressing concerns, and preventing surprises right before your trial is set to start. The FDA requires a briefing package with relevant materials and data at least a month before a requested meeting.
Stage 3: Clinical Development
In the early stages of clinical development, the investigational focus is safety monitoring.
- Phase 1 studies typically evaluate safety in a small number of healthy volunteers. Preliminary efficacy assessments in affected patients may be included in Phase 1 if the therapy is being evaluated in patients and is intended to address an unmet or life-threatening medical need.
- Phase 2 trials aim to determine whether the drug is safe and effective in a larger cohort of patients with the condition being treated. After this stage, an end-of-Phase 2 meeting with the FDA should be scheduled to review pivotal (Phase 3) trial design. The objective of the pivotal clinical trial is to demonstrate that the investigative drug has better efficacy than the established standard of care.
- Phase 3 trials assess whether the drug is safe and effective in a larger population of patients. If the data demonstrate this, the next step is to prepare a marketing application — either a new drug application (NDA) or a biologics license application (BLA) — for submission to the FDA. Similar to the IND, a pre-NDA/BLA meeting with the agency is useful for clarifying expectations and discussing application filing and format requirements.
Stage 4: Post-Market Safety Monitoring
Following marketing approval, the FDA requires post-market safety monitoring once products are made available to the public. Pharmacovigilance planning must outline actions to be taken when or if adverse events increase with drug uptake among larger numbers of patients. Phase 4 trials may be needed to further study the drug’s characteristics regarding safety, efficacy, expanded indications, and new formulations or routes of administration. These changes may result in extended market exclusivity under additional patent protection. Innovators also need to anticipate and plan for ongoing regulatory changes, such as labeling updates.
At Premier Consulting, A Division of Premier Research, we help sponsors achieve more rapid development and more efficient use of limited resources by integrating preclinical and clinical strategies and operations. Our consulting services provide access to a global network of product development and regulatory experts to help small biotech and specialty pharma companies design and execute comprehensive plans for product development, from early discovery through IND and post-approval lifecycle management. This allows you to keep product development moving forward efficiently while reducing risk and maximizing the opportunity for commercial success. To schedule a meeting with our team, click here.
[1] PhRMA. Biopharmaceutical Research & Development: The Process Behind New Medicines, 2015.