
Last Updated: January 7, 2026, 9 am UTC
Dose escalation studies are pivotal in the early phases of clinical drug development for evaluating the safety profile of a new therapeutic agent and identifying a recommended dose for further investigation. These studies lay the groundwork for understanding the pharmacokinetic and pharmacodynamic properties of novel therapies and, if designed and executed thoughtfully, can move seamlessly into phase 2 efficacy evaluations or even fast track into phase 3 registrational trials.
In this blog series, we explore tactics for optimizing phase 1 studies, strategies for successful early regulatory interactions, and considerations for enhancing the patient experience in a phase 1 setting.
Tactics for optimizing phase 1 studies
There are four key considerations when designing early phase clinical trials:
- Determine clear trial objectives. While phase 1 studies have traditionally focused on evaluating safety and pharmacologic activity, they can be designed to explore additional endpoints such as preliminary assessments of efficacy. For example, in oncology, tumor response signal may be included as a secondary, or even a primary, endpoint in phase 1 studies. Phase 1 studies can even be designed to examine the overall feasibility of therapy.
- Think through design challenges. Once the trial objectives have been determined, it is critical to work through any design challenges associated with those objectives. These challenges may include multiple endpoints, drug combinations, late-onset dose limiting toxicities (DLTs) predicted by preclinical data, or uncertainties in dose or schedule. Pinpointing the design challenges specific to a study will help the biostatistician design an appropriately-powered study.
- Define the study population. Narrowing in on the patient population most likely to benefit from the investigational therapy is essential. Consider whether disease stage, prior therapy, or biomarkers will influence the probability of success.
- Balance anticipated risks and potential benefit. Regulatory agencies will want some assurance that the potential benefit of a clinical trial outweighs the associated risk. Keep in mind that risk tolerance may vary based on the severity of the disease and the degree of unmet need.
Optimizing dose escalation study design
Common dose escalation designs include both standard rule-based designs, such as 3+3 or Rolling-6, and model-based designs (see Table 1).
Table 1. Comparison of dose escalation design types
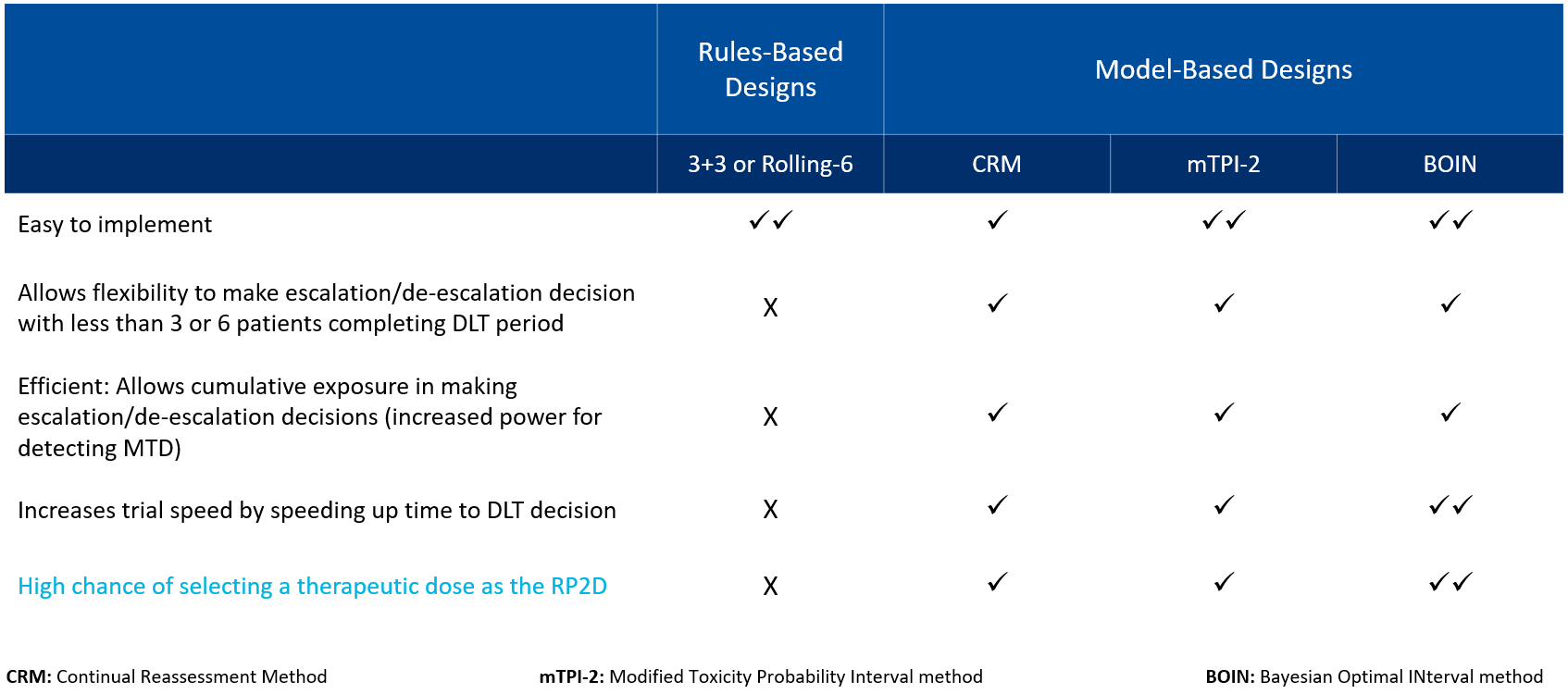
While rule-based designs are relatively easy to implement, they are associated with shortcomings due to their lack of flexibility in dose escalation and their limitations in assessing DLT. Model-based designs, on the other hand, allow flexibility in dose escalation or de-escalation decision-making and increase trial speed by accelerating the time to DLT. Most importantly, model-based designs such as the Bayesian Optimal Interval (BOIN) method increase the likelihood of selecting a therapeutically effective dose, rather than a maximum tolerate dose (MTD), as the recommended phase 2 dose (RP2D).
Streamlining dose escalation decision making
In dose escalation studies, Safety Review Committee (SRC) meetings serve as a critical mechanism for monitoring patient safety, evaluating the emerging safety profile of the investigational drug, and ensuring appropriate dose escalation. At Premier Research, we have developed a streamlined process that minimizes the time needed to make critical dose escalation decisions. As soon as the last patient in a cohort has been dosed, we work closely with the sites to get as clean as possible safety data that can be distributed to SRC members in advance of the meeting. Our goal is to have dose escalation decisions documented and investigators notified by the fifth day after the last patient dosed.
With a comprehensive understanding of key principles and strategies, researchers can navigate dose escalation studies with confidence, paving the way for successful clinical drug development and ultimately bringing new therapies to patients in need. Once the phase 1 study has been designed, the next step is to get buy-in from the regulatory agencies on the proposed clinical trial.
Strategies for successful early regulatory interactions
Once the phase 1 study has been designed, the next step is to get buy-in from the regulatory agencies on the proposed clinical trial. To support pre-clinical meeting with the FDA or other regulatory bodies, it is useful to conduct an internal regulatory gap analysis. This analysis typically includes pharmacology, Good Laboratory Practices (GLP), non-clinical safety, and Good Manufacturing Practices (GMP) information that will be necessary for filing a Clinical Trial Application (CTA) or Investigational New Drug (IND) application. Any gaps identified will guide the additional work needed to resolve data limitations and deficiencies.
Where possible, sponsors are encouraged to take advantage of accelerated approval pathways. The FDA has instituted four programs intended to facilitate and expedite the development and review of new drugs to address unmet medical need:
- Fast track designation
- Breakthrough therapy designation
- Accelerated approval
- Priority review designation
The eligibility criteria for each of these programs can be found in the FDA guidance document, Expedited Programs for Serious Conditions | Drugs and Biologics. The FDA also offers a regenerative medicine advanced therapy (RMAT) designation.
The European Medicines Agency (EMA) has similar expedited approval programs in the form of PRIME and accelerated assessment of advanced therapy medicinal products (ATMPs).
While sponsors of early phase studies are often worry that early interaction with the FDA will increase regulatory scrutiny and requirements, informal meetings such as INTERACT and CATT can be extremely beneficial for getting early agency buy-in on the proposed development path and preparing for the pre-IND meeting.
Considerations for enhancing the patient experience
Increasingly, regulatory agencies and sponsors are emphasizing and embracing the importance of the patient experience in clinical trials. After all, the success of any trial relies upon the willingness of patients to participate. The clinical research industry as a whole is undergoing a paradigm shift where patients are not just study subjects, but also collaborators.
Sponsors have traditionally engaged investigators to review their study protocols. Now, in addition to investigator review, sponsors are seeking feedback from patients and their caregivers to better understand the disease journey and the complexity of their burden. This patient-centric view looks at the clinical trial protocol through the lense of those who are living with the condition and shifts the focus from inclusion/exclusion criteria and assessments to outcomes that are meaningful to patients and their families (see Figure 1).
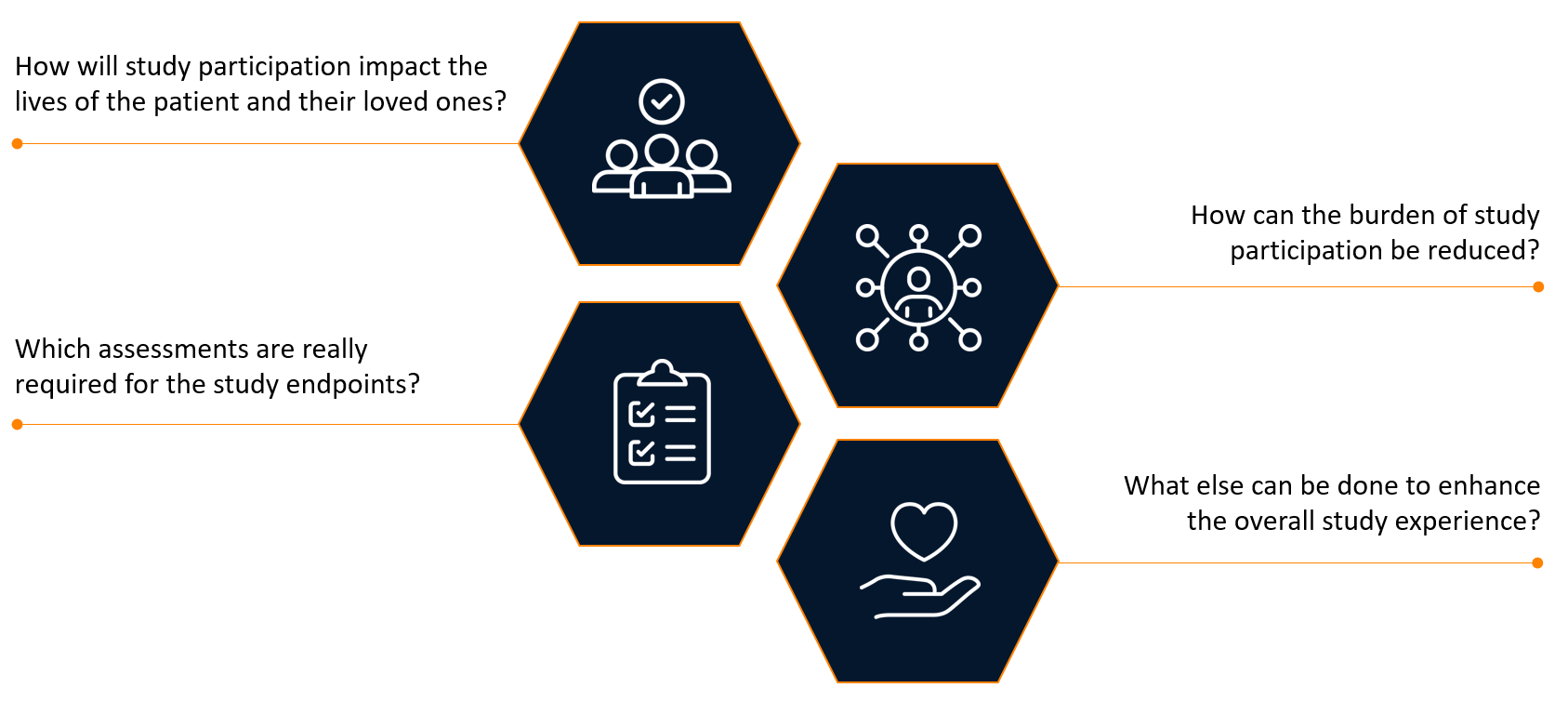
Figure 1. Assessing the patient centricity of a clinical trial
Patient centricity may be particularly important in phase 1 studies, where patients are enrolling for altruistic reasons rather than for an expectation of significant benefit. Ensuring that these patients have a positive experience increases the likelihood that they will enroll in other clinical trials upon progression or recommend clinical trial participation to another patient. Too often, strategies for patient retention only surface after a study is ongoing and may require protocol amendments. Instead, implementing patient-focused strategies from the initial stages of study planning can increase enrollment and retention, improve compliance, and increase patient satisfaction and engagement, all of which lead to better clinical trial outcomes and data quality.
Designing a patient-centric trial
When designing a patient-centric phase 1 trial, consider the following:
- Incorporate patient perspectives, preferences, and priorities.
- Analyze whether assessments are necessary or nice to have.
- Minimize patient burden.
- Leverage digital tools and remote monitoring technologies.
- Select endpoints that have high clinical relevance to improve accuracy.
- Include outcomes that are tailored to patients.
- Consider patient-meaningful endpoints that yield patient-relevant outcomes.
Conclusion
Dose escalation studies are the critical first step in clinical drug development and getting these studies right can help to accelerate later phase trials. At Premier Research we understand how to optimize early phase protocol development, navigate the pre-IND process, facilitate adaptive trial design and execution, and incorporate strategies for patient centricity to support enrollment and retention.
To learn more about how we can help you master dose escalation studies, contact us.
 Webinar
Webinar 


 Perspectives Blog
Perspectives Blog 