As scientific knowledge, clinical experience, and acceptance of gene therapy products have evolved, so have the regulatory frameworks for ensuring the safety of these novel treatments. To date, there is no harmonized international standard for regulating gene therapy products; however, the U.S., EU, and Japan have established regulatory frameworks with subtle variations. Understanding how gene therapy products are regulated — and how to navigate regional or national regulatory differences — can help you develop an efficient product development plan to bring your treatment to patients in need.
In this blog post, we review the regulatory frameworks for gene therapies in the U.S., EU, and Japan, with a focus on programs designed to streamline the product development and approval process.
Regulatory authorities with gene therapy oversight
When a regulatory authority has delineated a framework for the development of gene therapies, that framework often also applies to cell and tissue therapies. Given the broad scope of these frameworks, the regulatory authorities in the U.S., EU, and Japan have established committees or divisions that specialize in the evaluation of gene therapies:
- U.S. – Within the U.S. Food and Drug Administration and its Center for Biologics Evaluation and Research (CBER), there is an Office of Tissues and Advanced Therapies
- EU – In addition to the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use, there is a specialized Committee for Advanced Therapies that covers gene therapies
- Japan – Under the Pharmaceuticals and Medical Devices Agency (PMDA) and the Ministry of Health, Labor, and Welfare, there is an Office of Cellular and Tissue-based Products
The established frameworks for gene therapies in these three countries include expedited programs for facilitating the development, review, and registration of cell and gene therapy products.
Programs to promote product development
Each of these countries offers programs that help streamline the development of gene therapies. In the U.S., there are three programs designed to promote product development — fast track, breakthrough therapy, and Regenerative Medicine Advanced Therapy (RMAT) designations. As fast track designation was the first to be developed, many of the features of this designation are incorporated into the breakthrough therapy and RMAT designations as well. The EU offers Priority Medicines (PRIME) designation, and Japan offers the Sakigake designation.
A common thread to all the designations above is a requirement that the product under development be used to treat a serious condition associated with severe morbidity for which there is an unmet medical need (see Figure 1). The language of the criteria varies from designation to designation, and the requirement for unmet medical need may be indirectly stated as a requirement that the product under development demonstrates improved efficacy or safety compared to currently available therapies. Another common element of all these designations is that they can be lost if circumstances change and the product no longer meets the criteria for designation.
Of note, fast track in the U.S., PRIME in the EU, and Sakigake in Japan allow for the use of non-clinical data, either alone (for fast track) or in conjunction with clinical data (PRIME and Sakigake), to support the designation request.
Figure 1. Overview of programs to promote gene therapy development
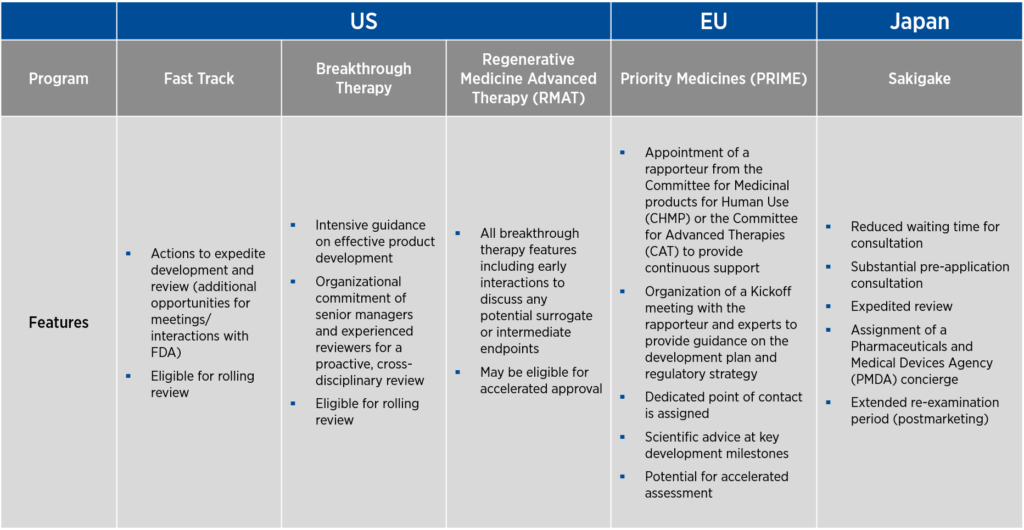
Sponsors who attain these designations benefit from increased access to — and senior-management-level feedback from — the regulatory authority that grants the designation. Through frequent meetings, the sponsor and the regulatory authority can achieve alignment on study design and data requirements.
You will also benefit from increased flexibility, including the ability to submit components of the biological license application (BLA) for review before submission.
Programs to accelerate application review
If a product proceeds successfully through clinical development, all three countries offer programs to expedite the review of BLAs or marketing applications. In the U.S. and Japan, these programs are called priority review, and in the EU, the program is known as accelerated assessment. As with the programs that promote product development, these expedited review programs have specific requirements and features (see Figure 2).
Figure 2. Overview of expedited review programs for gene therapy
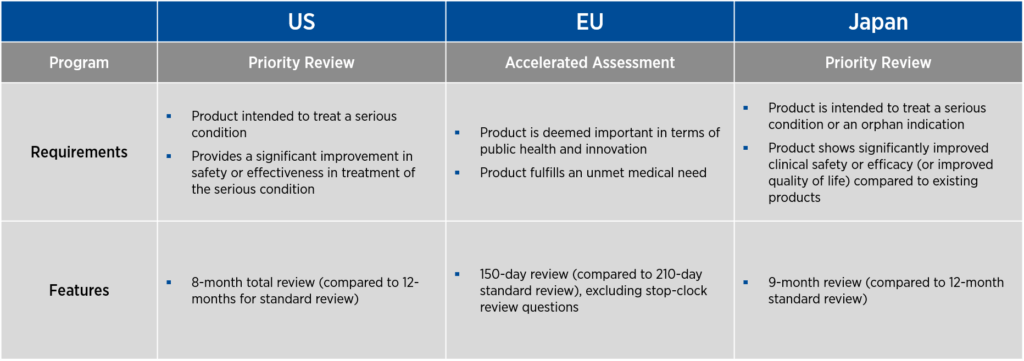
In the U.S., priority review offers an eight-month total review period, compared to 12 months for standard review. In the EU, accelerated assessment provides a 150-day review period, excluding stop-clock review questions, compared to the usual 210-day standard review. In Japan, priority review is associated with a nine-month review, compared to the standard 12-month review period.
Programs to expedite registration
Conditional approval is a mechanism for expediting the registration pathway of a promising therapy. Each country utilizes different terminology for conditional approval:
- U.S. – Accelerated approval
- EU – Conditional marketing authorization
- Japan – Conditional and term-limited approval
With these expedited registration pathways, it is possible that you may only need to conduct the first of two pivotal trials or be able to use a surrogate endpoint as the efficacy endpoint for your pivotal study in order to receive conditional approval. A key component of any expedited registration pathway is that confirmatory studies need to be conducted in a timely manner after conditional approval has been granted.
There are rare situations where marketing authorization is granted without confirmatory studies in the EU and Japan. In the EU, this scenario is known as marketing authorization under exceptional circumstances. Once granted, this type of marketing authorization can last up to five years, but its status is revisited once every year. In Japan, conditional approval without a confirmatory study is known as the conditional early approval system.
An evolving landscape
A relatively new development in the U.S. is the FDA’s Initial Targeted Engagement for Regulatory Advice on CBER products (INTERACT) meeting, an informal, non-binding, one-hour consultation that enables you to obtain preliminary advice for innovative investigational products on issues that are not yet at the pre-IND meeting phase. The INTERACT program is intended for products that “introduce unique challenges due to the unknown safety profiles resulting from the use of complex manufacturing technologies, development of innovative devices, or cutting-edge testing methodologies.”1
For more information on the existing regulatory frameworks for gene therapy, watch our webinar, Achieving the Promise of Gene Therapy: New Pathways to Overcome Patient Enrollment and Safety Challenges.
Key takeaways
As regulatory authorities continue to issue new guidance or publish resources that assist with the interpretation of the regulations, it is critical for sponsors to stay current with evolving standards and best practices. If you have questions about gene therapy regulations or eligibility for expedited programs, contact us to schedule a conversation.
1 U.S. Food and Drug Administration. INTERACT Meetings. FDA website. Accessed October 19, 2020.