To develop a successful gene therapy, sponsors must conduct research studies that balance the need for safety oversight and robust clinical evidence with the challenges of finding the right patients, pairing them with qualified sites, providing informed consent, and confirming they are capable of — and committed to — fulfilling all protocol requirements.
In this second part of a three-part blog series on gene therapy development, we explore the key considerations for designing gene therapy trials.
Considerations for early phase gene therapy trials
Gene therapy products are distinct from other types of pharmaceutical products and their unique characteristics may impact study design. First, gene therapy products may be associated with risks such as the potential for prolonged biological activity, immunogenicity, or off-target effects. Administration of these treatments may also involve invasive procedures. Moreover, preclinical data are not always informative as it is not always feasible to conduct traditional preclinical pharmacokinetic studies.
Given these product characteristics, the design of a gene therapy trial must consider safety procedures or controlling immunogenicity and use of chemistry, manufacturing, and controls data to assess appropriate safety and dosing regimens. Study design must also address ethical concerns. For instance, when using an AAV-based therapy with a known immunogenic response, current thinking is that each patient may only be able to receive a single dose of that AAV serotype. Patients participating in a first in human dose escalation study are likely to receive an ineffective dose level and may be ineligible to receive another dose.
Elements of study design
Figure 1. Overview of gene therapy study design
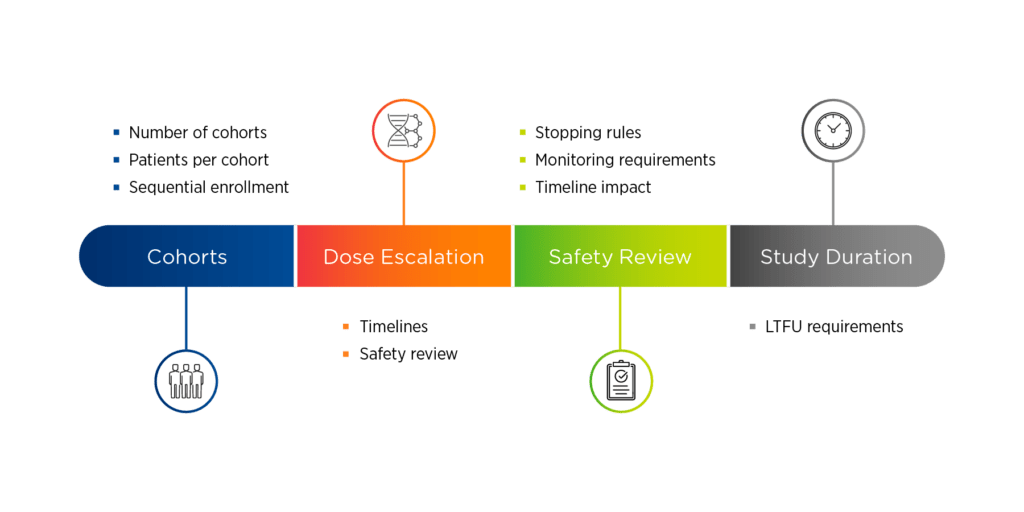
Four key elements of gene therapy study design are:
1. Cohorts. Sponsors will need to determine the number of cohorts, the number of patients per cohort, and how those patients will be enrolled. Sequential enrollment, where only one patient is enrolled at a time, is most common in first in human gene therapy trials. Another issue to consider is the use of placebo control, which the FDA may favor but the EMA may not. One option is to use a control group, which can be achieved by randomizing patients into a dose delayed run-in period so they can serve as their own comparator.
2. Dose escalation. If there is dose escalation, there will need to be a timeline for safety reviews. These safety reviews can either occur between individual patients or between cohorts. A common study design in Phase 1/2 gene therapy trials is dose escalation followed by dose expansion.
3. Safety reviews. Frequent safety reviews are inherent in gene therapy studies and will be accompanied by stopping rules and monitoring requirements, which may have an impact on study timelines.
4. Study duration. Sponsors will need to understand the long-term follow-up requirements associated with their candidate therapy.
Study design must also incorporate the role of the data safety committee, which serves as an impartial mediator that objectively evaluates patient data to assess safety, and sometimes efficacy, before the study can move forward.
Vector considerations
The type of vector used will impact both cell transport and the need for immunosuppression. AAV-based gene therapies are genetically modified organisms that will likely require cold chain transport. Drug stability must also be considered when scheduling patient screening visits. Historically, immunosuppressants were not always given with AAV-based gene therapies, especially if administered into the central nervous systems. Today, however, immunosuppressants are seeing increased used with all types of in vivo AAV therapy due to the possibility that drug-related immune responses might render the drug inactive before it could have its desired effect. Typically, immunosuppressants are given prior to gene therapy administration to dampen the immune response to the vector and then tapered after dosing.
For autologous cell transfer, such as with lentiviral vectors, cells can be modified either locally in the hospital or centrally in a manufacturing facility. Chain of identity is critical and having processes in place to ensure that cells or containers of cells are clearly marked with patient identification is essential for avoiding mix ups. Chain of custody must also be confirmed with a log of who is responsible for the cells at each step of the manufacturing process. In addition, it is important to understand and define transport requirements, whether cells are transported live and cryopreserved for increased stability. The need for immunosuppressants with ex vivo gene therapies will depend on the immunogenicity of the surface proteins. Chemotherapy is commonly used as a conditioning agent, but if immunogenic surface proteins are present, an immunosuppressant may be added.
If immunosuppressant therapy is indicated, it is important to monitor compliance. Sponsors may consider using an electronic diary device, as non-compliance with immunosuppressant therapy not only represents a safety concern, but also puts data quality at risk.
Protocol review
Involving key stakeholders in protocol review can help minimize the need for protocol amendments, which can be costly and time-consuming. Asking key opinion leaders to review the protocol and getting their buy-in on the types and timing of proposed assessments is critical. They may identify an issue with an endpoint or an aspect of study design that was overlooked. Patient advocacy groups are another valuable resource, as they can review the protocol from the perspective of patients and their families and serve as a potential resource for study participants. Finally, engaging with regulatory agencies early and often can help sponsors identify and address potential issues or concerns with the protocol.
In part three of this blog series, we discuss strategies for selecting the right gene therapy sites and optimizing patient enrollment and engagement.
At Premier, we have conducted more than 60 cell and gene therapy studies in the past five years, combining our significant experience in viral vectors with deep therapeutic expertise in oncology, rare disease, and pediatric research. Click here to learn more about how Premier can help move gene therapy research forward.