Last Updated: January 5, 2026, 2 pm UTC
Development of liquid biopsies for early cancer detection requires careful planning. Understanding the regulatory environment and the challenges of conducting the studies needed for approval is integral to success. Here, we review the regulatory pathways for in vitro diagnostics (IVDs) in both the US and the EU and offer strategies for designing and operationalizing large-scale validation studies of liquid biopsies intended for cancer screening.
Regulatory pathways: Where risk and clinical decisions intersect
In the US, there are multiple regulatory pathways for IVDs, including laboratory-developed tests (LDTs), 510(k), and pre-market approval (PMA). The existing scheme is based on risk—the higher the risk, the tighter the controls or the threshold for performance required.
LDTs are IVDs that are designed, manufactured, and used within a single laboratory. Laboratories that run samples, whether collected at home, in a lab or in a clinical care setting, need to be accredited by Clinical Laboratory Improvement Amendments (CLIA) accrediting agencies. Currently, there is proposed regulation that would create a new risk-based framework for both LDTs and IVDs. Another consideration for LDTs is that many payers, whether public or private, will expect accreditation to ISO 15189:2022, Medical laboratories — Requirements for quality and competence, when the device developer is not registered with FDA or a Notified Body in the EU.
The 510(k) pathway is used for class II devices and submission involves proving substantial equivalence to a predicate device. As a rule, the 510(k) path is for those tests that have established performance, for instance, blood typing and certain biomarkers.
PMA is required for class III devices that are higher risk or that represent new technologies with no predicate and submission involves proving that the device is safe and effective for the end user. Generally, liquid biopsies for early cancer detection will need to follow the PMA pathway. However, FDA also has a special submission type called breakthrough device designation (BDD) for products that have the potential to provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions.1 Due to their pan-indication potential and general ease of sample acquisition, liquid biopsies for early cancer detection have advantages that may make them eligible for BDD. Devices that receive this designation enter the Breakthrough Devices Program, which offers the benefits of expedited priority review and timely access to FDA reviewers and scientists.
In the EU under the In Vitro Diagnostics Regulation (IVDR), all IVDs must be classified according to both patient and public health risk. Classification follows seven rules, which are used to place an IVD into one of four risk classes (see Table 1). Each risk class requires its own path to conformity.
Table 1. IVD regulatory pathways in the EU
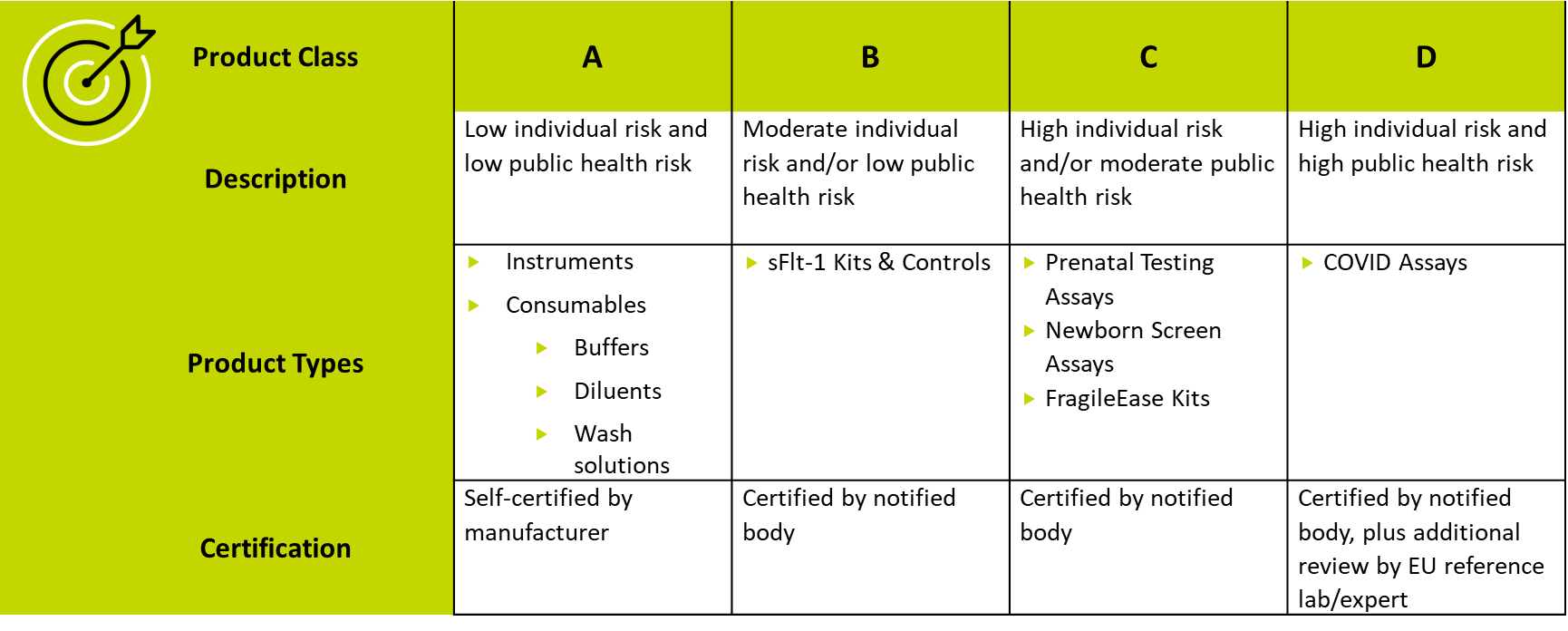
For a screening claim, the highest risk to an individual is a false negative. This is the harm that regulatory bodies focus on, so liquid biopsies for early cancer detection generally fall into Class C or D and require certification by a notified body and possibly additional review by a reference laboratory or expert.
How to confirm the performance of your liquid biopsy test
To obtain regulatory approval for a liquid biopsy test, developers generally need to demonstrate scientific validity, analytical validity, clinical validity, and clinical utility. Together, these characteristics constitute the performance of the test.
- Scientific validity is the association of an analyte with a clinical condition or a physiological state.
- Analytical validity is the ability of the test to correctly detect or measure that analyte.
- Clinical validity is the ability of the test to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user.
- Clinical utility is the capacity of the test to improve cancer diagnosis, treatment, management, or prevention.
The analytical and clinical validation studies required to support a liquid biopsy test will depend on the intended use and indications for use. Analytical validation can be challenging because obtaining enough clinical samples harboring a known targeted mutation for repeat testing can be difficult. Clinical validity can also be challenging for a screening test because of the large numbers of samples required to confirm true positives and true negatives and to determine the rates of false positives and false negatives.
Developers planning to conduct clinical performance studies should carefully study the requirements of ISO 20916, In vitro diagnostic medical devices — Clinical performance studies using specimens from human subjects — Good study practice. There are two types of clinical performance studies:
- Non-interventional studies, where test results do not influence patient management and treatment decisions.
- Interventional studies, where test results may influence patient management and treatment decisions. Whether a liquid biopsy study is considered interventional may depend on the existence of a standard of care screening procedure to serve as a final clinical comparator. Interventional studies where investigational test results are strictly used only for patient management require an Investigational Device Exemption (IDE) from FDA.
Designing clinical validation studies: A formulaic approach
Most clinical validation studies for early cancer detection tests follow a similar design: Samples are collected and run, yielding a qualitative result, and then a clinical procedure is performed to confirm both positives and negatives. When designing the study, it is important to consider:
- Retrospective vs. prospective sampling. If using biobank samples, collect samples that will confirm performance in the intended use population and ensure that those samples come from an average risk population that would meet the anticipated inclusion and exclusion criteria for the clinical study. Research has shown that running samples that have been identified or collected retrospectively often results in performance that is higher than it would have been in the real world.
- Sample size calculation. How many samples will be needed to reach statistical significance for a screening claim? Powering will be based on sensitivity.
- Inclusion and exclusion criteria. These should align with the existing screening paradigm for that indication, if applicable. Importantly, for a screening claim, participants must be asymptomatic.
- Standard of care. If there is an existing screening paradigm, the study protocol should follow standard of care. For example, for breast cancer, if the liquid biopsy suggests cancer and the confirmatory mammogram reveals a BI-RADS finding, the protocol should call for repeat imaging in 6-12 months, not biopsy.
Other issues to address include how to confirm true negatives, how to characterize precancerous lesions, and whether long-term follow-up is needed. Some studies have used full-body DEXA scans as a confirmatory procedure, but these scans are very expensive, making them less feasible for larger studies.
Operationalizing large-scale clinical validation studies: Tips for success
Large-scale clinical validation studies are an expensive undertaking, so planning for success is essential.
Clinical validation plan. The first step is a clinical validation plan that confirms performance thresholds and acceptance, establishes primary and secondary objectives and endpoints, and defines eligibility criteria for both subjects and samples. At this point, engaging with regulators and performing a gap analysis is extremely useful for ensuring that the study is on the right track.
Site identification and qualification. Potential sites should be evaluated for feasibility based on their research infrastructure, understanding of the patient enrollment pathway, and access to the right patient population.
Sample management. Clinical validation studies involve many thousands of samples, so having a robust process and standard operating procedures (SOPs) for managing these samples—from collection and transport to processing and storage—is crucial. It is also important to identify all the data elements—including demographic and physiological variables—that will need to be tracked over the course of the study.
Ideally, study samples will be collected, frozen, and stored, and then run in a batch after design lock to maximize accuracy and consistency. To do this, it may be necessary to perform bench studies, if they have not already been done, to determine a stability window for the analyte. In general, cell-free DNA (cfDNA) can be stored for long periods.
Pilot study. In some cases, it may be advisable to conduct a pilot study in the intended use population to generate initial validation data and assess any performance changes in the real world. An added benefit of a pilot study is streamlining start-up for the large-scale study as sites will already be up and running.
Recruitment and engagement. Recruitment can be optimized through strategic selection of geographic areas where incidence of the cancer of interest are higher or through age-weighted enrollment skewed toward older populations. Decentralized recruitment—through pharmacies or labs and especially for self-collection tests—can also help to boost recruitment. Another factor to consider in recruitment is diversification. In addition to being an FDA priority, diversity is especially important for screening, as minorities tend to have higher rates of many cancers.
Some studies may involve long follow-up periods for confirming clinical results, so maximizing patient and site engagement is essential. Retention efforts should focus both on short-term compliance—for example, screening mammography following a positive early breast cancer detection test result—and longer-term adherence to any follow-up procedures required to confirm the clinical result. Staying engaged with participants through IRB-approved thank you notes or reminder cards, or study updates, can also help to improve retention.
Risk-based monitoring. Gone are the days of conducting 100% onsite source data verification (SDV) for IVD studies. Instead, sponsors should base their monitoring plans on the most critical data elements, such as human subject protection and data integrity around the clinical result, to reduce the time and costs associated with routine onsite visits to all clinical sites. Technology is critical to the success of the entire risk-based monitoring process.
Your regulatory pathway will determine your operational needs
The development process for in vitro diagnostic devices is complex, particularly for liquid biopsy tests intended for screening and early cancer detection. Early, frequent communication with regulators ensures selection of an appropriate pathway to approval, and proactive planning enables developers to operationalize the large-scale—and, at times, long-term—clinical validation studies required to successfully traverse that path.
To see how we’ve successfully managed large-scale clinical validation studies in the past, read our case study here.
[1] US Food and Drug Administration. Breakthrough Devices Program: Guidance of Industry and Staff, December 18, 2018.
 Webinar
Webinar 


 Perspectives Blog
Perspectives Blog 