Transdermal and topical delivery systems (TDS) are important dosage forms that allow delivery of a drug to local tissue or provide systemic delivery through the skin. These drug products provide a number of advantages for patients, but can be challenging to develop. In November 2019, the FDA issued a draft guidance entitled Transdermal and Topical Delivery Systems – Product Development and Quality Considerations: Guidance for Industry.[1] The document provides the FDA’s current thinking on product design, pharmaceutical development, product quality, and the performance and safety evaluations for TDS.
Why develop a TDS product?
TDS have a number of advantages over other routes of administration. First, TDS can improve patient compliance, especially in patients with swallowing difficulties or gastrointestinal (GI) irritation. Convenience and ease of use are other key reasons for TDS development. From a drug exposure perspective, systemic absorption via TDS (through the skin) avoids potential enzymatic digestion or drug hydrolysis in the acidic stomach environment. It also avoids first pass metabolism, which can provide greater systemic levels of certain active ingredients. Additionally, due to the controlled release mechanisms of TDS, the duration of drug exposure can be designed in some cases to be prolonged for multiple days.
TDS – Development Considerations
When developing a TDS product, a number of considerations should be evaluated at the early stages of product development:
- What is the skin permeation capability of the active ingredient? Is it safe to use on the skin?
- Does the TDS formulation have the potential to irritate or sensitize the skin?
- What will be the duration of the TDS application? Where will the product be applied? Will the application site need to be rotated?
- Are the drug levels needed for efficacy well characterized? Can a sufficient dose be incorporated into the TDS?
- Can a reasonably sized patch be developed for the indication and target population?
Two main types of TDS are the reservoir and matrix style, distinguished by how the active ingredient is incorporated into the product. A matrix-style TDS product contains the active ingredient dissolved or suspended in a matrix containing adhesive, penetration enhancers, preservatives and other excipients.
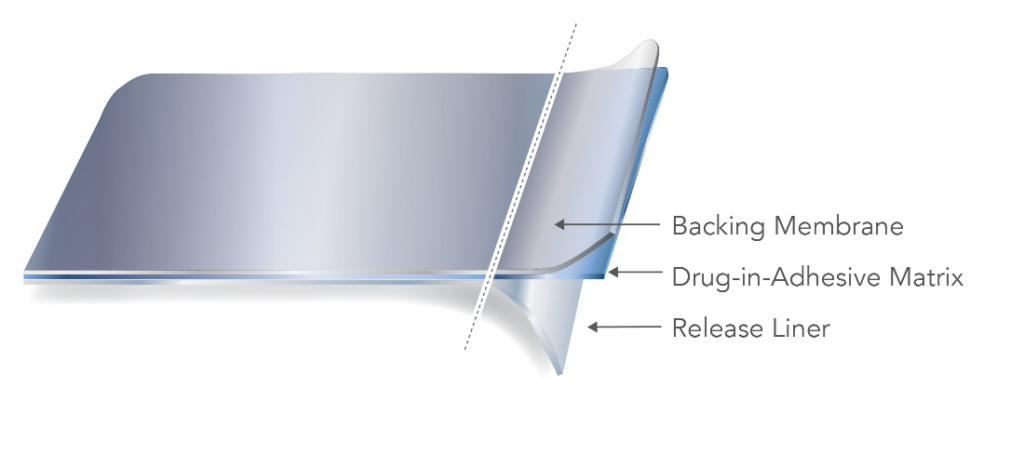
A reservoir TDS product contains the drug in a heat-sealed area between the backing membrane and a semi-permeable membrane that is in contact with the skin.
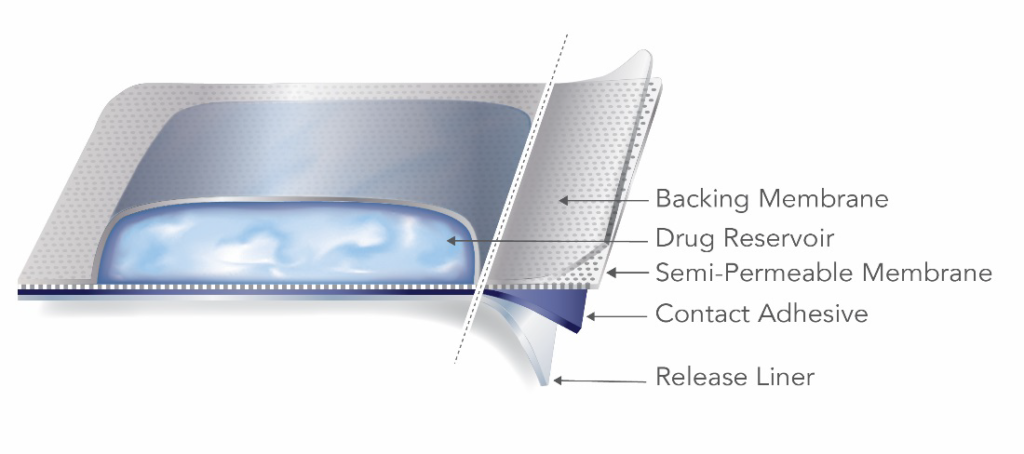
The reservoir TDS design has inherent failure potential and safety risks. Strong pressure applied to a reservoir TDS can cause the product to rupture, releasing the contents of the drug-containing reservoir in an uncontrolled manner. For this reason, the FDA advises manufacturers and applicants to focus development efforts on matrix-style TDS. The guidance further states that “[a]pplicants are strongly encouraged to consult the Office of Pharmaceutical Quality early in the development process prior to pursuing a reservoir design.”
Regulatory Considerations for TDS
From a regulatory perspective, TDS products are combination products and are required to comply with 21 CFR part 4 subpart A (Current Good Manufacturing Practice [CGMP] Requirements for Combination Products).[2] This describes how 21 CFR parts 210 and 211 (drug CGMPs) and 21 CFR part 820 (device Quality System regulation) apply to combination products. Additionally, developers of TDS should be aware of the design control regulations outlined in 21 CFR part 820.30, to confirm there are no negative interactions between the product’s constituent parts and to make sure the combined use results in a combination product that is safe, is effective and performs as expected.
The Quality Target Product Profile (QTPP)
In the guidance, the FDA recommends that before initiating the development of a TDS product, applicants should establish a Quality Target Product Profile (QTPP), outlining the quality characteristics of the product and taking into account its safety and efficacy. The QTPP may include the following:
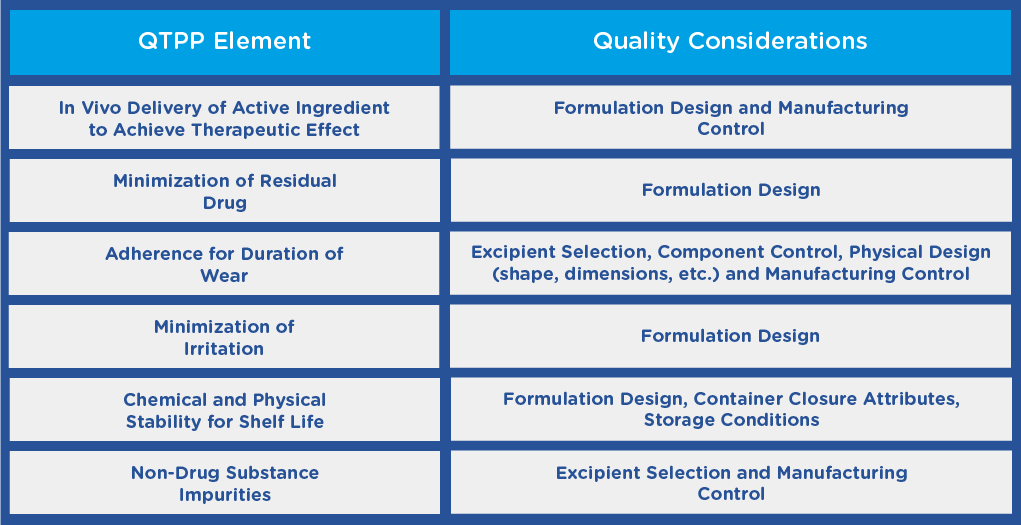
Additional QTPP elements may exist depending on the indication, or other functional requirements, such as the size of the finished product, which may depend on the location on the body that the TDS product is to be applied.
Critical Quality Attributes
Early in product development, applicants should establish a list of critical quality attributes (CQAs). Using the QTPP as a guide, applicants should outline the CQAs that define the physical, chemical, biological or microbiological properties or characteristics that should be within an appropriate limit, range or distribution to ensure the desired product quality. This list of CQAs will likely evolve over the product development lifecycle, as additional information is obtained.
The FDA guidance summarizes typical CQAs for the drug substance, drug product and excipients for TDS. Notable for TDS are the qualification of adhesives; the guidance outlines parameters for evaluation of adhesive polymers as raw materials, adhesive as a laminate (in the absence of the Active Pharmaceutical Ingredient [API] and other excipients), and adhesive in the final product (along with drug substance and other excipients and components).
Another important attribute of the TDS is the identifying information printed on the product itself. This should be established early in development to ensure that the labeling process or inks used for printing do not interact with the TDS, and to assess inks in extractable/leachable studies. Applicants should be aware that products that are clear or colored to match human skin tone can make it difficult to find the TDS on the patient; this can lead to potential medication errors (for example, if the product becomes unknowingly detached or through the application of multiple products). Therefore, it is recommended that the backing membrane is printed with ink of adequate contrast for easy identification.
Information to be Submitted in an Application
The new guidance outlines the quality information to submit in a marketing application for a TDS product. The guidance describes information to include in the pharmaceutical development portion of the application, including expectations for registration and exhibit batches. For TDS, registration/exhibit batches should be manufactured from three distinct laminates, each made using different lots of API, adhesives, backing and/or other critical elements of the product. Release and stability sampling should be representative of the full length and width of the laminates to demonstrate robust manufacturing. Since TDS products are sensitive to small differences in the manufacturing process, a table comparing the clinical, BE, registration/exhibit and proposed commercial batches should be included in the application. For each batch, the table should specify the manufacturing process used, results of critical in-process tests, yield and reconciliation data.
The guidance also describes the product characterization studies, manufacturing information and control evaluations to be completed for TDS products when preparing a marketing application. This includes evaluations of crystallization potential of the formulation, residual drug assessment, the effects of heat on drug delivery and adhesion testing. Both in vitro and in vivo adhesion testing should be performed, with in vivo assessments representative of real-world use.
Conclusions and Recommendations
The development of TDS is important, as there are many patients that benefit from these products. The new draft guidance provides a much-needed perspective on the FDA’s expectations for the development of these products, and how applicants should develop and properly characterize these products for a marketing application. In addition to the information provided in the new guidance, meeting with the FDA early in the development program is recommended and will help clarify the nonclinical, clinical, quality and regulatory requirements specific to a given program.
Is your company considering developing a TDS product? The experts at Regulatory Professionals (RPI), A Division of Premier Research, have assisted in the development of TDS products through all stages and can provide guidance and expertise for your program. For more information, contact us.
[1] Center for Drug Evaluation and Research. (2019, November). Transdermal and Topical Delivery Systems – Product Development and Qua. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/transdermal-and-topical-delivery-systems-product-development-and-quality-considerations
[2] Center for Drug Evaluation and Research. (2017, January). Current Good Manufacturing Practice Requirements for Combination Products. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/current-good-manufacturing-practice-requirements-combination-products
 Perspectives Blog
Perspectives Blog